Ein Zellkern oder Nukleus (lat. nucleus = Kern, gr. karyon = Kern) ist ein im Zytoplasma gelegenes, meist rundliches Organell der
eukaryotischen Zelle, das den Großteil des Erbguts enthält.
- Allgemeine Merkmale:
- Ca. 7 µm groß und macht 10–25 % des Zellvolumens aus
- Hauptmerkmal der Eukaryoten (Prokaryoten wie
Bakterien und Archaeen besitzen keinen abgegrenzten
Zellkern) - Enthält fast die gesamte DNA der Zelle (mit Ausnahme
der mitochondrialen DNA) - DNA liegt als Chromatin in zwei Zuständen vor:
- Euchromatin: Locker gepackte doppelsträngige DNA, transkriptionsaktiv, im Lichtmikroskop hell sichtbar
- Heterochromatin: Dicht gepackt, inaktiver Bereich, im Lichtmikroskop dunkel sichtbar
- Prozesse innerhalb des Zellkerns:
- DNA-Replikation: Verdopplung des Erbguts während der S-Phase des Zellzyklus
- Transkription: Synthese von RNA (mRNA, tRNA, rRNA, snRNA) aus DNA-Vorlage
- RNA-Prozessierung: Modifikation der prä-mRNA (Capping, Spleißen, Polyadenylierung)
- Zusammenlagerung von Ribosomenvorstufen im Nukleolus (rRNA + ribosomale Proteine)
- Kernmembranen:
- Der Zellkern ist von einer Doppelmembran (innere und äußere Kernmembran) umgeben
- Die äußere Membran ist mit dem rauen endoplasmatischen Retikulum (rER) verbunden
- Der perinukleäre Raum zwischen beiden Membranen steht mit dem Lumen des rER in Verbindung
- An der inneren Kernmembran liegt die Kernlamina (Lamina fibrosa nuclei), bestehend aus Lamin A, B, C (Intermediärfilamente)
→ Mechanische Stabilität und Anker für Chromatin
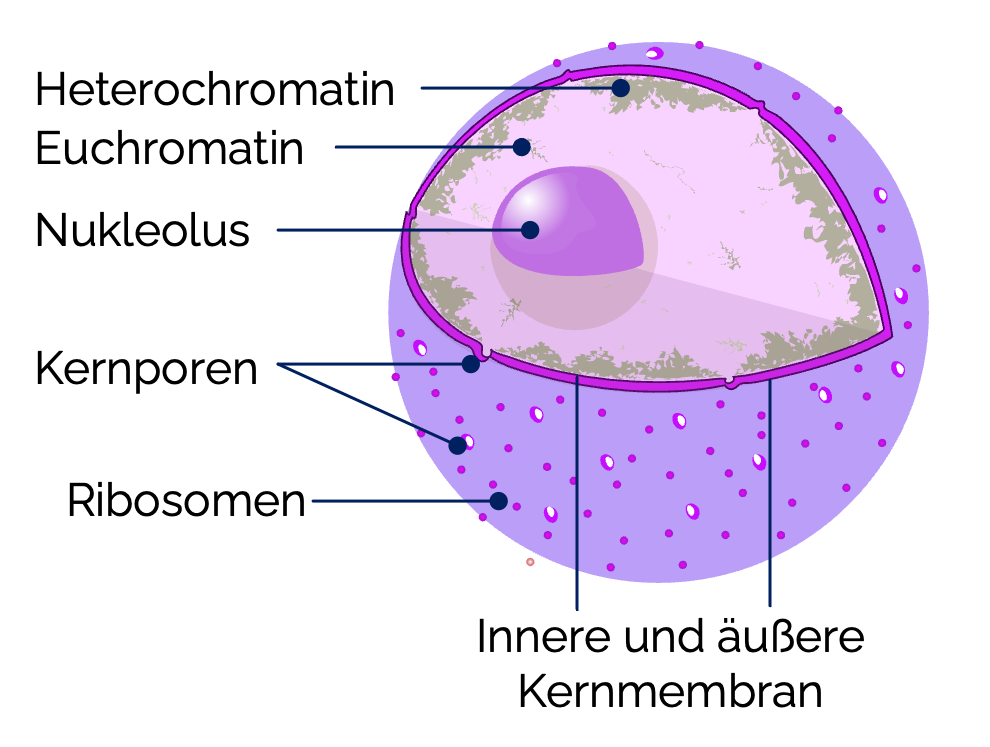
Kernporen
- Die Kernmembran wird durch Kernporen durchbrochen wird. Sie entstehen durch
- Fusion beider Kernmembranen
- Regulieren den Transport von Molekülen zwischen Zytoplasma und Karyoplasma
- Passiv: kleine Moleküle (<5 kDa) können frei diffundieren
- Aktiv: größere Moleküle benötigen Karyopherine (Transportrezeptoren). Die Karyopherine erkennen die Kernlokalisierungssequenz (NLS) oder die Kernexportsequenz (NES) bestimmter Moleküle (v.a. Proteine) und transportieren das Protein durch die Kernporen
- Der Import (Zytoplasma → Karyoplasma) z.B. von DNA-Polymerasen oder Laminen geschieht über Importine
- Der Export (Karyoplasma → Zytoplasma) z.B. von mRNA oder tRNA geschieht über Exportine
- Der aktive Transport ist energieabhängig und wird durch die GTPase Ran gesteuert
Nucleoli
Innerhalb des Zellkerns befinden sich eine oder mehrere rundliche Strukturen, die als Nukleoli (Kernkörperchen) bezeichnet werden. Im Lichtmikroskop erscheinen sie als deutlich erkennbare, dunkle basophile Bereiche. Sie treten besonders ausgeprägt in Zellen mit hoher Proteinbiosynthese auf, beispielsweise in Leberzellen, Nervenzellen, Tumorzellen).
- Aufbau:
- Umgeben von perinukleärem Heterochromatin
- Nicht membranumschlossen
- Enthalten Abschnitte der DNA, die rRNAs (ribosomale RNAs) codieren
- rRNA-Gene liegen in den Nukleolus-Organisator-Regionen (NORs) der akrozentrischen Chromosomen (13, 14, 15, 21, 22)
- Funktionen:
- Transkription der rRNA-Gene
- Zusammenbau mit ribosomalen Vorläuferproteinen
- Export dieser Vorstufen ins Zytoplasma über Kernporen

Das endoplasmatische Retikulum (ER) ist ein membranöses System, das die Zelle durchzieht und aus vesikulären und tubulären Strukturen besteht. Es ist direkt mit der äußeren Membran des Zellkerns verbunden und kann in Lamellen, Zisternen und Tubuli unterteilt werden.
Raues endoplasmatisches Retikulum (rER):
Das raue endoplasmatische Retikulum (rER) ist mit Ribosomen besetzt und ist der Ort der Synthese von Proteinen für die Zellmembran, Lysosomen und Sekretion. In Nervenzellen stellen die Nissl-Schollen ein raues ER dar.
- Funktionen:
- Ribosomen docken an das rER an und setzen die Proteinsynthese fort
- Bildung von Sekretproteinen, Membranproteinen und Proteinen für das ER oder den Golgi-Apparat
- Transport der Syntheseprodukte in Vesikeln zum Golgi-Apparat oder direkt zum Bestimmungsort
- Vesikel zum Golgi-Apparat werden mit auf der Proteinhülle mit dem Coat protein Complex II ausgestattet
- Erste posttranslationale Modifikation von Proteinen im rER (z. B. N- Glykosylierung, Faltung)
- Ribosomen docken an das rER an und setzen die Proteinsynthese fort
Glattes endoplasmatisches Retikulum (sER):
Das glatte endoplasmatische Retikulum (englisch smooth endoplasmic reticulum, sER) hat keine Ribosomen und ist der Ort der Synthese von Lipiden, Steroidhormonen und Entgiftung toxischer Substanzen über Cytochrom-P450- Enzyme. In der quergestreiften Muskulatur dient das sER als Calciumspeicher. In Hepatozyten ist das sER an der Gluconeogenese und der Glykogenolyse beteiligt, da das membrangebundene Enzym Glukose-6-Phosphatase Glukose-6-Phosphat in freie Glukose umwandelt. Aus dem glatten ER schnüren sich zusätzlich Vorläufervesikel von Peroxisomen ab.
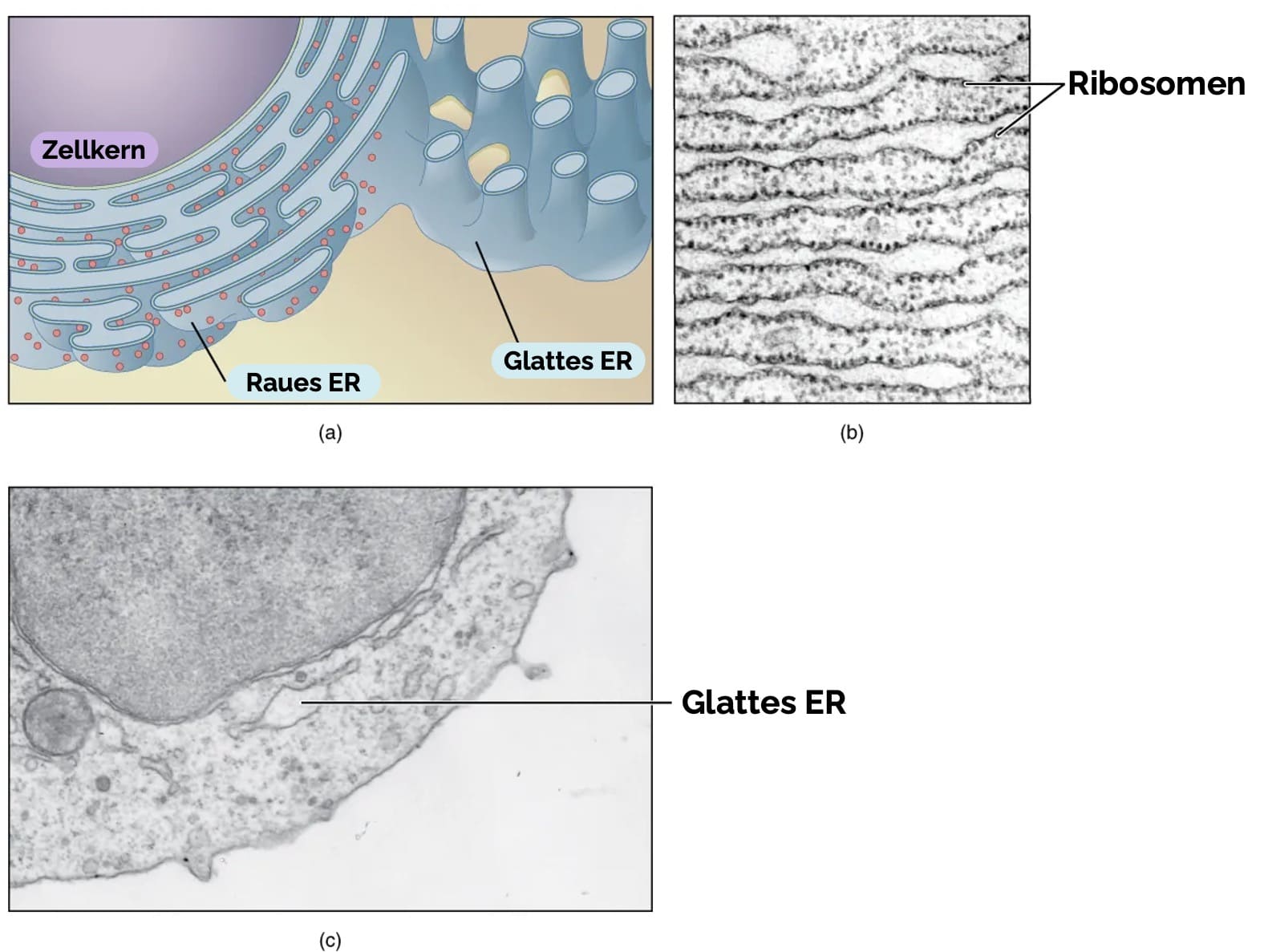
Ribosomen sind essentielle Organellen, die in Pro- und Eukaryoten vorkommen und eine wichtige Rolle bei der Translation spielen.
- Ribosomen sind membranlos und bestehen aus einer großen und einer kleinen Untereinheit
- Die Untereinheiten bestehen aus Ribonucleinsäuren und Proteinen.
- Ribosomen können frei im Zytoplasma vorkommen oder an das raue endoplasmatische Retikulum gebunden sein
- Sie dienen der Synthese von Proteinen durch Übertragung der Nucleotidsequenz der mRNA in die Aminosäuresequenz eines Proteins
- Die von Ribosomen hergestellten Proteine können im Zytoplasma verbleiben oder in die Mitochondrien, den Zellkern oder Peroxisomen transportiert werden
- Mehrere Ribosomen können gleichzeitig an ein mRNA-Molekül gebunden sein und bilden ein Polysom
- Die Biosynthese der Ribosomen findet im Zellkern statt, in dem bereits vorhandene Ribosomen spontan Proteine für neue Ribosomen herstellen

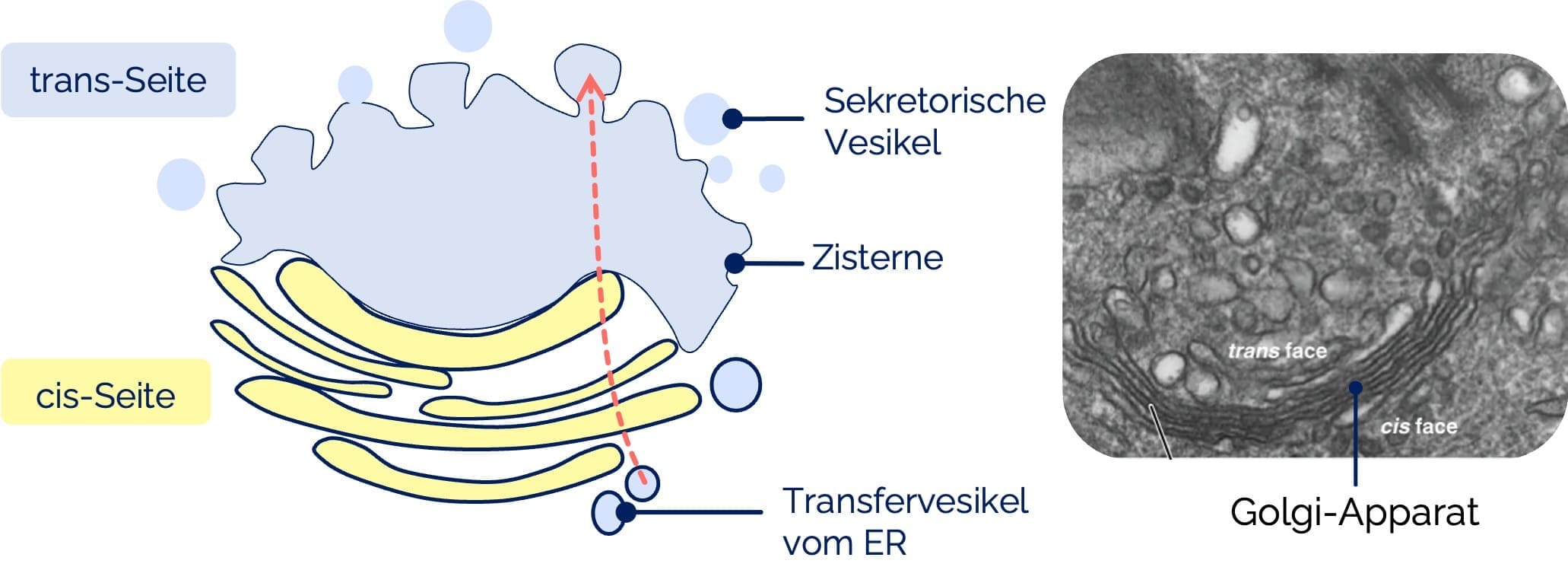
Der Golgi-Apparat ist ein membranöses Organell, das eine zentrale Rolle bei der Sortierung, Modifikation und dem Weitertransport von Proteinen und Lipiden spielt.
- Aufbau:
- Besteht aus flachen Membranstapeln (Zisternen), die funktionell in verschiedene Bereiche gegliedert sind:
- Cis-Golgi-Netzwerk (cis-Golgi): Aufnahmeseite für ER-Vesikel
- Mediale Zisternen: Modifikationszone
- Trans-Golgi-Netzwerk (trans-Golgi): Sortier- und Exportseite
- Besteht aus flachen Membranstapeln (Zisternen), die funktionell in verschiedene Bereiche gegliedert sind:
- Funktion:
- Vesikel aus dem endoplasmatischen Retikulum (ER) fusionieren mit der cis-Seite und entlassen Proteine und Lipide in das Lumen der Golgi-Zisternen
- Das Material wird über Zisternenmigration oder Vesikeltransport durch den Apparat zur trans-Seite weitergeleitet
- Am trans-Golgi entstehen Vesikel, die entweder:
- Zum Export aus der Zelle bestimmt sind (Sekretproteine)
- In die Zellmembran eingebaut werden (Zellmembranproteine)
- Oder Enzyme für die Bildung primärer Lysosomen enthalten lysosomale Proteine
- Modifikationen im Golgi:
- Glykosylierung von Proteinen und Lipiden
- Sulfatierung von Glykoproteinen
- Acylierung
- Phosphorylierung von Mannoseresten (z. B. Mannose-6-Phosphat als Signal für lysosomale Enzyme)
- Klinische Relevanz: Fehlfunktionen des Golgi-Apparates können die Proteinsortierung und den Vesikeltransport stören und sind mit verschiedenen Erbkrankheiten und Stoffwechselstörungen assoziiert (z. B. kongenitale Glykosylierungsstörungen)
InfoGolgi-Apparat = Zellpost - sortiert und verpackt Proteine und Lipide für den Versand
Golgi-Apparat:
Membranumschlossene Organellen mit saurem pH (≈ 4,5), zuständig für den Abbau zelleigener und -fremder Strukturen.
- Funktionen:
- Abbau von Makromolekülen (Proteine, Lipide, Polysaccharide, Nukleinsäuren)
- Recycling von Zellbestandteilen (Autophagie)
- Abtötung phagozytierter Mikroorganismen
- Freisetzung lysosomaler Enzyme kann bei Schädigung die Apoptose (programmierter Zelltod) in Gang setzen
- Antigenprozessierung in Antigen-präsentierenden Zellen (APZ) → Beteiligung an der adaptiven Immunantwort durch MHC-II-Präsentation
- Entstehung:
- Primäre Lysosomen: Entstehen aus dem trans-Golgi-Netzwerk und enthalten noch inaktive lysosomale Enzyme
- Diese Enzyme werden am rauen endoplasmatischen Retikulum (rER) synthetisiert
- Im Golgi-Apparat erfolgt die Modifikation mit einem Mannose-6-phosphat- Rest, der als „Adressierungssignal“ für lysosomale Enzyme dient
- Anschließend werden die Enzyme als Vesikel am trans-Golgi abgeschnürt und liegen als primäre Lysosomen in der Zelle
- Sekundäre Lysosomen: Fusion von primären Lysosomen mit Endosomen (siehe Abbildung), Phagosomen oder Autophagosomen → Ort der Verdauung
- Saurer pH (~4,5) wird durch H+-ATPase erzeugt → aktiviert lysosomale Enzyme (saure Hydrolasen) → So wird sichergestellt, dass der enzymatische Abbau nur im Lysosom erfolgt. Es ist ein Schutzmechanismus, damit zelleigene Strukturen bei Leckage nicht geschädigt werden
- Enthalten zahlreiche saure Hydrolasen, z. B. Proteasen (spalten Proteine), Lipasen (spalten Lipide), Nukleasen (spalten DNA/RNA), Glykosidasen (spalten Zuckerketten), saure Phosphatasen
- Primäre Lysosomen: Entstehen aus dem trans-Golgi-Netzwerk und enthalten noch inaktive lysosomale Enzyme
- Klinische Relevanz: Lysosomale Speicherkrankheiten sind eine Gruppe seltener, genetisch bedingter Erkrankungen, bei denen aufgrund eines Enzymdefekts bestimmte Substrate in den Lysosomen nicht abgebaut werden können. Infolgedessen kommt es zur Akkumulation dieser Substanzen in verschiedenen Zelltypen, was zu zellulärer Dysfunktion und Organschäden führt (z.B. Morbus Gaucher, Tay-Sachs-Krankheit, Morbus Niemann-Pick)
MerkeDer histochemische Marker für Lysosomen ist die saure Phosphatase.

Peroxisomen sind kleine, runde Vesikel, die im Zytosol vorkommen. Sie oxidieren Aminosäuren und Fettsäuren unter Verbrauch von Sauerstoff.
- Aufbau und Vorkommen:
- Kleine, membranumhüllte Vesikel im Zytosol
- Entstehung: Abschnürung aus speziellem glattem ER
- Proteinimport: Aktiv aus dem Zytoplasma über spezifische Signalsequenzen
- Besonders zahlreich in Hepatozyten
- Funktionen:
- β-Oxidation sehr langkettiger Fettsäuren
- Abbau von Wasserstoffperoxid (H2O2) durch Katalase → Schutz vor oxidativem Stress
- Synthese von Etherlipiden (z. B. Plasmalogene)
- Beteiligung an der Gallensäuresynthese
- Mechanismus:
- Bei oxidativen Abbauprozessen entsteht Wasserstoffperoxid (H2O2)
- Katalase zerlegt H2O2 in H2O + O2, um Zellschädigung zu verhindern
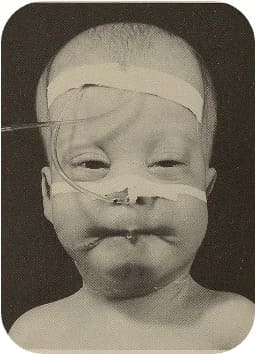
- Klinische Relevanz: Das Zellweger-Syndrom ist eine schwerwiegende, autosomal-rezessive Peroxisomen- Biogenesestörung, bei der Peroxisomen nur unzureichend oder gar nicht gebildet werden. Die Folge ist eine Akkumulation sehr langkettiger Fettsäuren und toxischer Metabolite, was zu schweren neurologischen, hepatischen und gastrointestinalen Symptomen führt. Die Erkrankung verläuft meist letal im ersten Lebensjahr
Säugling mit Zellweger-Syndrom:
Mitochondrien sind membranumhüllte Zellorganellen und spielen eine zentrale Rolle in der Energiegewinnung. Als „Kraftwerke der Zelle“ produzieren sie den Großteil des zellulären ATPs durch oxidative Phosphorylierung. Darüber hinaus sind sie an zahlreichen weiteren Stoffwechselwegen beteiligt und besitzen eine eigene DNA, was ihre evolutionäre Herkunft von endosymbiotischen Bakterien unterstreicht. Ihre Funktion ist essenziell für energieabhängige Prozesse in nahezu allen Geweben.
- Aufbau:
- Doppelmembran mit äußerer und innerer Membran
- Intermembranraum zwischen beiden Membranen, die Mitochondrien-Matrix befindet sich im Inneren
- Innere Membran:
- Einstülpungen: Cristae (Leistentyp), Tubuli (Röhrentyp; v. a. bei steroidbildenden Zellen), Sacculus-Typ (nur in der Zona fasciculata der Nebennierenrinde anzutreffen) und Prisma-Typ (in Astrozyten)
- Enthält kein Cholesterin, dafür das Phospholipid Cardiolipin (typisch für Mitochondrien)
- Mitochondrien enthalten eigene mitochondriale DNA (mtDNA) in der Matrix
- Ringförmige doppelsträngige DNA
- Nicht an Histone gebunden
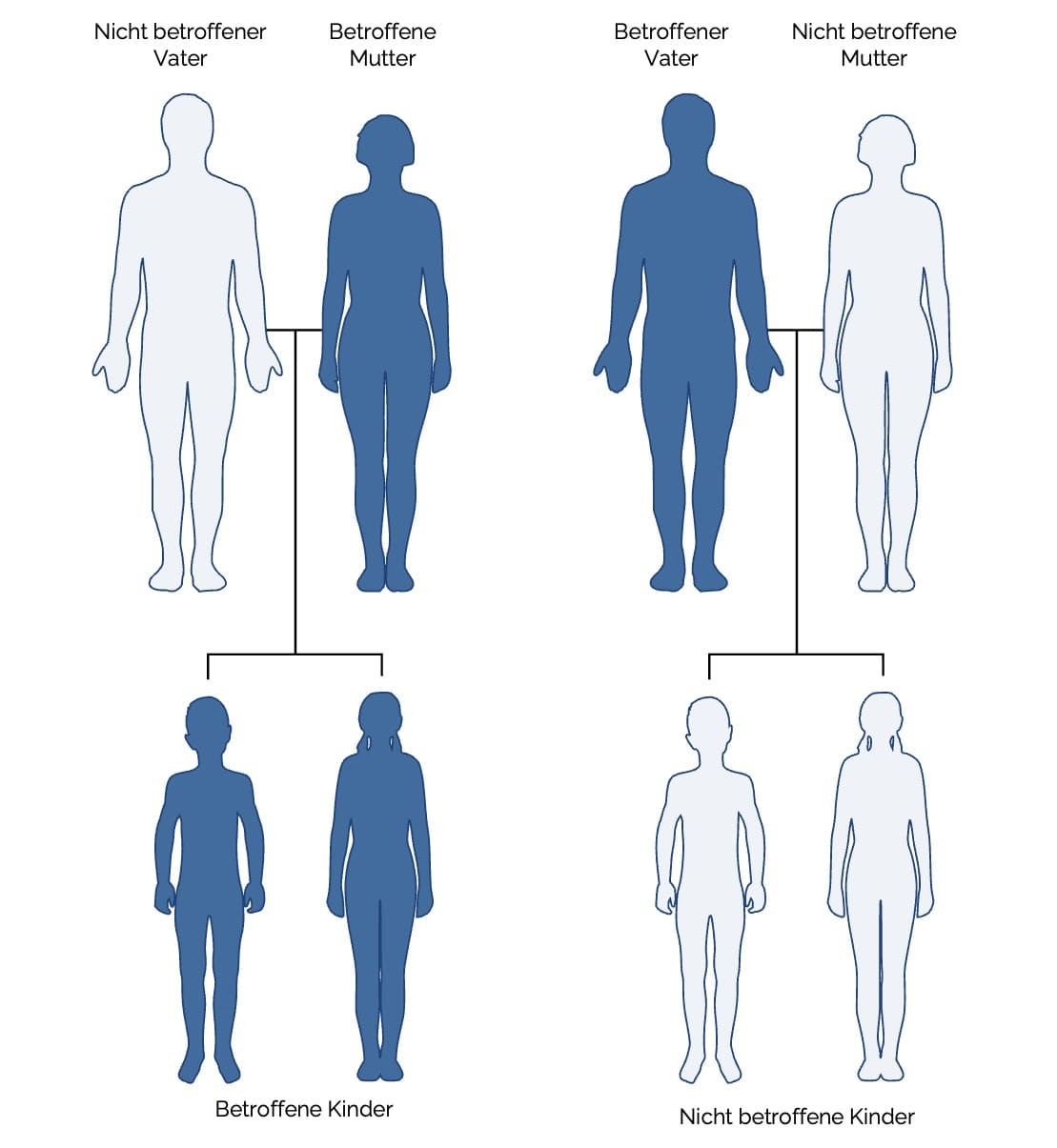
- Maternale Vererbung (väterliche Mitochondrien werden bei der Befruchtung nicht übertragen)
- Der Großteil der mtDNA besteht aus codierenden Sequenzen
- Über 90 % der mtDNA codiert für:
- Einige mitochondriale Proteine (z. B. Untereinheiten der Komplexe I, III, IV und V der Atmungskette)
- tRNA und rRNA
- Über 90 % der mtDNA codiert für:
- Die Transkription erfolgt durch eine mitochondriale RNA-Polymerase
- Die Translation findet an mitochondrialen Ribosomen statt
- Der Großteil der mitochondrialen Proteine werden jedoch im Zytoplasma synthetisiert
- Codierende DNA liegt im Zellkern
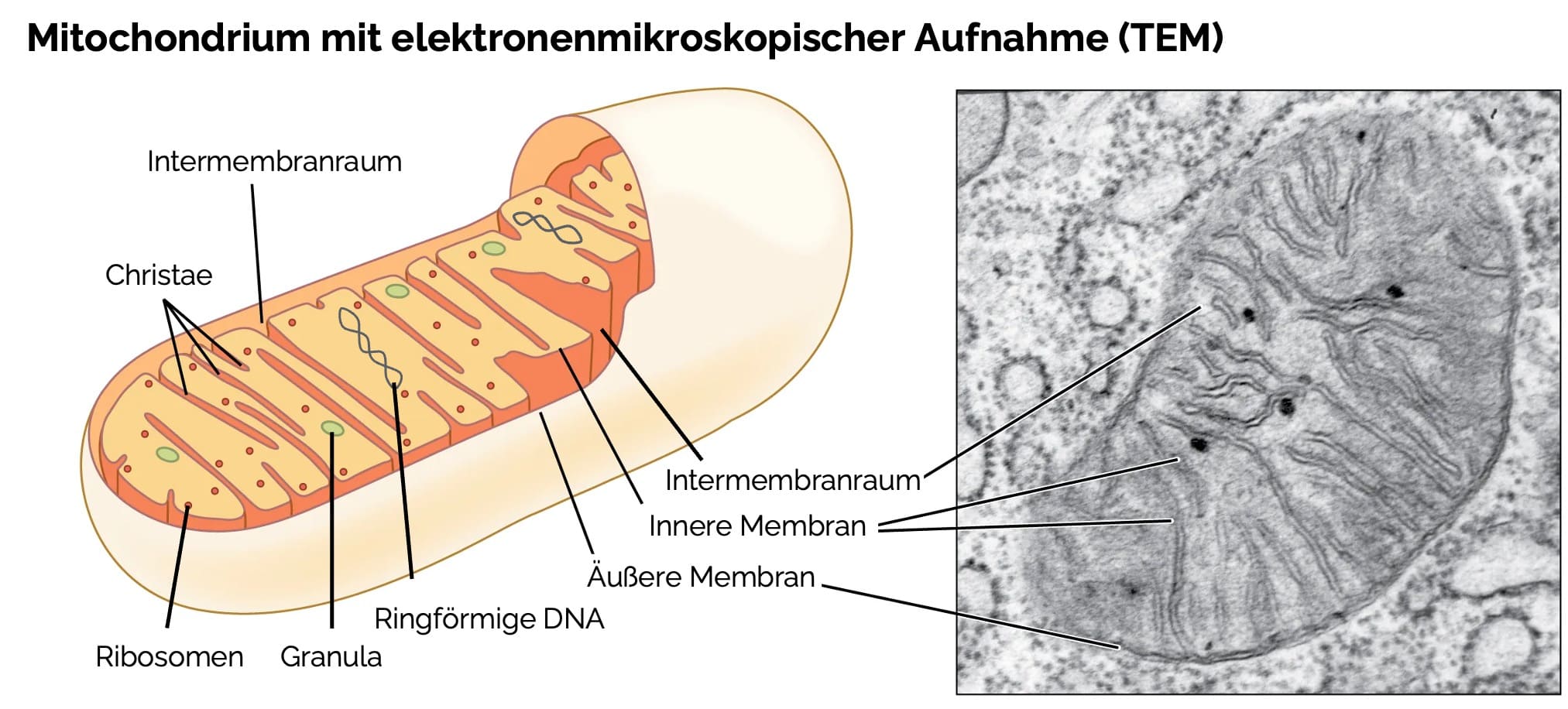
- Funktionen:
- ATP-Synthese über oxidative Phosphorylierung
- Zentrale Stoffwechselprozesse:
- Citratzyklus
- β-Oxidation (Fettsäureabbau)
- Teile der Gluconeogenese
- Speicherung von Calcium
- Regulation der Apoptose
- Klinische Relevanz: Mitochondriopathien sind Erkrankungen durch Störungen der mitochondrialen Funktion → v. a. muskuläre, neurologische und metabolische Symptome. Bei Mitochondriopathien mit mtDNA-Mutation sind ausschließlich Nachkommen betroffener Mütter gefährdet, da Spermien bei der Befruchtung keine Mitochondrien übertragen.